 内分泌科17-α羟化酶缺陷症病例分析专题报告.docx
内分泌科17-α羟化酶缺陷症病例分析专题报告.docx
《内分泌科17-α羟化酶缺陷症病例分析专题报告.docx》由会员分享,可在线阅读,更多相关《内分泌科17-α羟化酶缺陷症病例分析专题报告.docx(8页珍藏版)》请在优知文库上搜索。
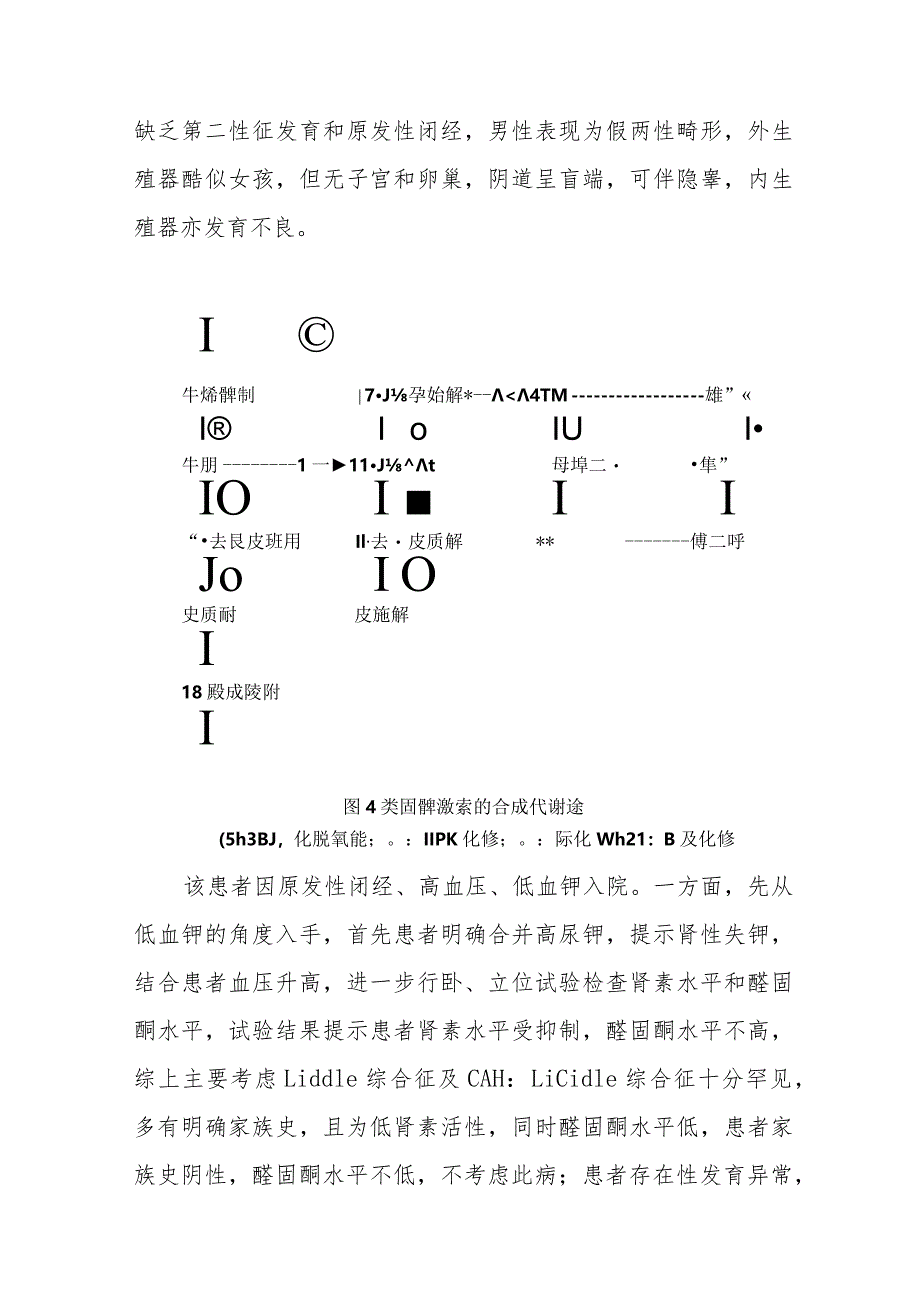
1、内分泌科17羟化酶缺陷症病例分析专题报告知识回顾及病例分析先天性肾上腺皮质增生症(Congenitaladrenalhypoplasia,CAH)是一组常染色体隐性遗传性代谢疾病,其主要病理机制在于肾上腺皮质类固酮激素生物合成过程中某种代谢酶的先天性缺陷,引起肾上腺皮质激素合成不足,经下丘脑-垂体-肾上腺轴反馈调节,使促肾上腺皮质激素释放激素(corticotropin-releasinghormone,CRH)及促肾上腺皮质激素(adrenocorticotropichormone,ACTH)分泌增加(图4),导致肾上腺皮质增生及代谢紊乱。涉及CAH的代谢酶缺陷中,17。-羟化酶缺陷症属非常
2、少见的类型,由XYP17A基因突变所致,可引起肾上腺皮质醇合成不足,反馈刺激ACTH分泌增加,使肾上腺盐皮质激素合成增加皮质酮和脱氧皮质酮(DOC),可达正常的3060倍。本病患者一般没有肾上腺皮质功能减退表现,这是由于该酶缺陷时皮质酮分泌大量增加,而皮质酮本身具有一定程度的糖皮质激素活性。由于皮质醇和性激素合成受阻,而DOe和皮质酮分泌增多,导致临床主要表现为高血压、低血钾、碱中毒及性发育缺陷,女性患者表现为性幼稚、青春期缺乏第二性征发育和原发性闭经,男性表现为假两性畸形,外生殖器酷似女孩,但无子宫和卵巢,阴道呈盲端,可伴隐睾,内生殖器亦发育不良。I牛烯髀制7J孕始解*-17至夫2$ZS*1
3、7-%17.20fiMExlTTC%F53O分17*等分11.CxlMTTCTY645S5%17a.iWCxlUUC-KiUCF93CW174JB10%17.20t8MElCIT96W11-B17.20WMMEx2TYS106P17-B17,200UUCYUC“14VVaFlkBh.20ittJGAUYUUDU6V911MVe17.20&祭CiCGCACABKI葡丽子码Stopisr17-H11.20MMI43GAG-TAGStop194170超S17203”bc4M518入469ZB17-4UI17.20bc4CGArA&StoP2”17-fiHOI17.20KtUiAGG-UGCHM7C斗
4、分%-*aD200”1x7UUC-UGCM17C17454tExS移4个AIMC分ITa-XiUI17.X)BHMEtCGCTaR496C17aJSJI11),hRl针对17-羟化酶缺陷症,最主要的治疗是激素替代治疗,包括糖皮质激素和性激素。糖皮质激素替代治疗的目的在于补充肾上腺皮质醇分泌的不足,抑制ACTH的过量分泌,减少DOC等物质的分泌,使血压下降并纠正低血钾。该患者行中剂量地塞米松抑制试验,ACTH可抑制至1.1pmol/L以下,反映良好,遂予地塞米松治疗。适量的雌激素可增加骨骼的钙盐沉积、加速骨髓闭合,增加骨量,减少骨丢失。该患者已至中年,应补充雌激素维持其社会性别,还有利于改善其骨




- 配套讲稿:
如PPT文件的首页显示word图标,表示该PPT已包含配套word讲稿。双击word图标可打开word文档。
- 特殊限制:
部分文档作品中含有的国旗、国徽等图片,仅作为作品整体效果示例展示,禁止商用。设计者仅对作品中独创性部分享有著作权。
- 关 键 词:
- 内分泌 17 羟化酶 缺陷 病例 分析 专题报告
 优知文库所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
优知文库所有资源均是用户自行上传分享,仅供网友学习交流,未经上传用户书面授权,请勿作他用。
 2022自身免疫性肝炎的管理JSH临床实践指南主要内容.docx
2022自身免疫性肝炎的管理JSH临床实践指南主要内容.docx
-
2022迟发性医源性胆道损伤的内镜外科治疗策略(全文).docx
-
2022药物性肝损伤的管理的分类、诊断和肝活检(第一部分).docx
-
2022骨质疏松性椎体压缩骨折的诊治现状(全文).docx
-
2022降低糖尿病风险饮食建议(全文).docx
-
2022非肝硬化性门静脉高压症的临床管理现状(全文).docx
-
2022骨质疏松的非药物治疗策略(全文).docx
-
2022震颤的分类、病因、治疗(全文).docx
-
2022血小板在肝细胞癌发生发展中的作用(全文).docx
-
2022软产道裂伤致隐匿性产后出血的早期识别及处理(全文).docx
-
CDC指南:单纯性VVC主要内容.docx
-
2022间质性膀胱炎膀胱疼痛综合征诊治(全文).docx
-
2022近端胃切除术双通道重建研究进展(全文).docx
-
C-反应蛋白(CRP)指标临床应用价值.docx
-
2022褪黑素在女性不孕相关疾病中的生殖调节研究(全文).docx
-
2022高危妊娠滋养细胞肿瘤的治疗(全文).docx
-
2022骨盆投射角与髋关节疾病关系的研究进展(全文).docx
-
2022髋关节置换手术入路的选择(全文).docx
-
CRP、hs-CRP、WBC的相互关系解析.docx
-
2022骨塑建在骨质疏松症防治中的作用(全文).docx
-
2022银行员工个人工作心得体会范文(五篇).docx
-
XX养老机构节能降耗实施方案.docx
-
2022季学期班级安全工作计划表范文(五篇).docx
-
2022党风廉政专题党课讲稿范文(通用三篇).docx
-
2022BMJ痛风的诊断和治疗(全文).docx
-
2022ESMO胃癌指南新疗法(全文).docx
-
2022ROSAH综合征的临床特点与治疗(全文).docx
-
2022最新版中国胃癌诊疗指南解读(全文).docx
-
《电工电子》考试大纲.docx
-
《战略合作框架协议》.docx
-
XX市参加重大体育比赛奖励办法(征求意见稿).docx
-
伙委会会议流程.docx
-
在全市城市基层党建引领基层治理工作会议上的发言.docx
-
财收表16-“发展运行性费用”细化预算表.docx
-
第五次全国经济普查统计重点业务综合培训大会上的讲话.docx
-
物业公司维序队长考核评分表.docx
-
集美大学横向项目成员变更申请表.docx
-
集体土地使用权调查表.docx
-
第二次主题教育党支部党员深入个人检视问题清单及整改措施.docx
-
雨课堂一体化智慧教学平台身份绑定操作说明.docx
-
物业公司消防演练.docx
-
财务经费审批权限授权委托书.docx
-
在全市决战四季度大干一百天动员部署会上的交流发言 .docx
-
在全市宣传思想文化系统调研成果汇报交流会上的讲话.docx
-
雷电防护装置竣工验收审批事项服务指南.docx
-
财政支出项目绩效评价自评报告.docx
-
第四届中国创新挑战赛宜宾技术创新需求调查表.docx
-
物业公司节能降耗措施大全.docx
-
在全市基层治理模式创新工作观摩会上的交流发言.docx
-
物业公司财务部员工岗位职能.docx
-
简洁范文网络研修心得体会多篇.docx
 手足口病健康教育宣传要点.docx
广西流动儿童在居住地享有关爱服务基础清单.docx
宽坪小学食堂食品卫生安全应急预案(精选19篇).docx
卫生健康信息数据元目录 第17部分:卫生健康管理(代替WS 363.17—2011).docx
卫生健康信息数据元目录 第12部分:计划与干预(代替WS 363.12—2011).docx
卫生健康信息数据元值域代码第7部分:体格检查(代替WS 364.7—2011).docx
卫生健康信息数据元值域代码 第2部分:标识(代替WS 364.2—2011).docx
卫生健康信息数据元值域代码第14部分:卫生健康机构(代替WS 364.14—2011).docx
卫生健康信息数据元目录 第11部分:医学评估(代替WS 363.11—2011).docx
卫生健康信息数据元值域代码第6部分:主诉与症状(代替WS 364.6—2011).docx
手足口病健康教育宣传要点.docx
广西流动儿童在居住地享有关爱服务基础清单.docx
宽坪小学食堂食品卫生安全应急预案(精选19篇).docx
卫生健康信息数据元目录 第17部分:卫生健康管理(代替WS 363.17—2011).docx
卫生健康信息数据元目录 第12部分:计划与干预(代替WS 363.12—2011).docx
卫生健康信息数据元值域代码第7部分:体格检查(代替WS 364.7—2011).docx
卫生健康信息数据元值域代码 第2部分:标识(代替WS 364.2—2011).docx
卫生健康信息数据元值域代码第14部分:卫生健康机构(代替WS 364.14—2011).docx
卫生健康信息数据元目录 第11部分:医学评估(代替WS 363.11—2011).docx
卫生健康信息数据元值域代码第6部分:主诉与症状(代替WS 364.6—2011).docx